Single-cell RNA expression profiling of ACE2, the putative receptor of Wuhan 2019-nCov
Abstract
A novel coronavirus (2019-nCov) was identified in Wuhan, Hubei Province, China in December of 2019. This new coronavirus has resulted in thousands of cases of lethal disease in China, with additional patients being identified in a rapidly growing number internationally. 2019-nCov was reported to share the same receptor, Angiotensin-converting enzyme 2 (ACE2), with SARS-Cov. Here based on the public database and the state-of-the-art single-cell RNA-Seq technique, we analyzed the ACE2 RNA expression profile in the normal human lungs. The result indicates that the ACE2 virus receptor expression is concentrated in a small population of type II alveolar cells (AT2). Surprisingly, we found that this population of ACE2-expressing AT2 also highly expressed many other genes that positively regulating viral reproduction and transmission. A comparison between eight individual samples demonstrated that the Asian male one has an extremely large number of ACE2-expressing cells in the lung. This study provides a biological background for the epidemic investigation of the 2019-nCov infection disease, and could be informative for future anti-ACE2 therapeutic strategy development.
Severe infection by 2019-nCov could result in acute respiratory distress syndrome (ARDS) and sepsis, causing death in approximately 15% of infected individuals1,2. Once contacted with the human airway, the spike proteins of this virus can associate with the surface receptors of sensitive cells, which mediated the entrance of the virus into target cells for further replication. Recently, Xu et.al., modeled the spike protein to identify the receptor for 2019-nCov, and indicated that Angiotensin-converting enzyme 2 (ACE2) could be the receptor for this virus3. ACE2 is previously known as the receptor for SARS-Cov and NL634–6. According to their modeling, although the binding strength between 2019-nCov and ACE2 is weaker than that between SARS-Cov and ACE2, it is still much higher than the threshold required for virus infection. Zhou et. al. conducted virus infectivity studies and showed that ACE2 is essential for 2019-nCov to enter HeLa cells7. These data indicated that ACE2 is likely to be the receptor for 2019-nCov.
The expression and distribution of the receptor decide the route of virus infection and the route of infection has a major implication for understanding the pathogenesis and designing therapeutic strategies. Previous studies have investigated the RNA expression of ACE2 in 72 human tissues8. However, the lung is a complex organ with multiple types of cells, and such real-time PCR RNA profiling is based on bulk tissue analysis with no way to elucidate the ACE2 expression in each type of cell in the human lung. The ACE2 protein level is also investigated by immunostaining in lung and other organs8,9. These studies showed that in normal human lung, ACE2 is mainly expressed by type II and type I alveolar epithelial cells. Endothelial cells were also reported to be ACE2 positive. However, immunostaining analysis is known for its lack of signal specificity, and accurate quantification is also another challenge for such analysis.
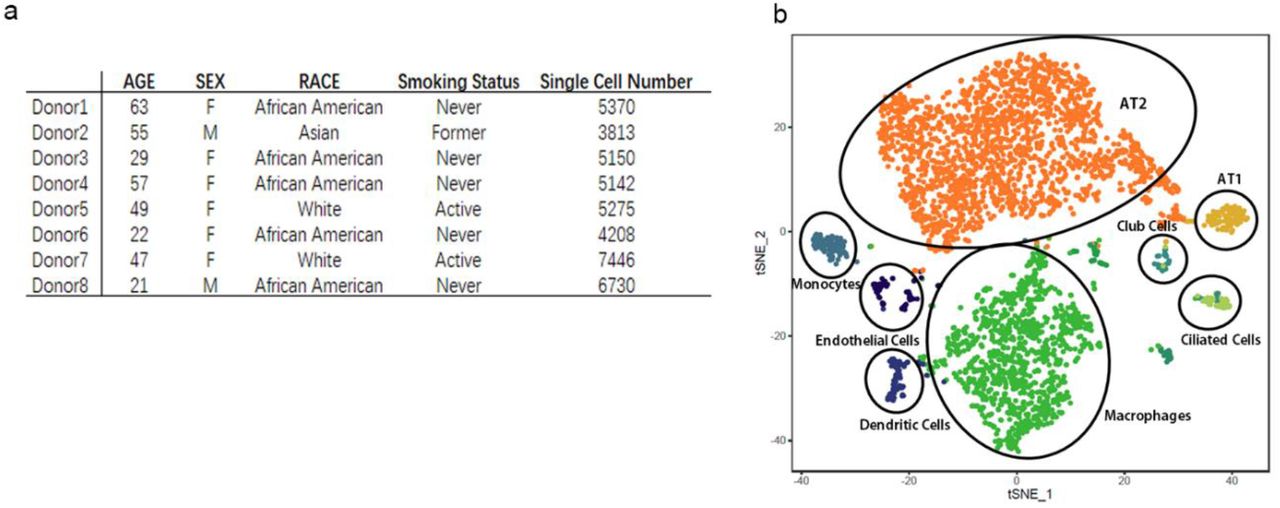
The recently developed single-cell RNA sequencing (scRNA-Seq) technology enables us to study the ACE2 expression in each cell type and give quantitative information at single-cell resolution. Previous work has built up the online database for scRNA-Seq analysis of 8 normal human lung transplant donors10. In current work, we used the updated bioinformatics tools to analyze the data. In total, we analyzed 43,134 cells derived from normal lung tissue of 8 adult donors. We performed unsupervised graph-based clustering (Seurat version 2.3.4) and for each individual, we identified 8~11 transcriptionally distinct cell clusters based on their marker gene expression profile. Typically the clusters include type II alveolar cells (AT2), type I alveolar cells (AT1), airway epithelial cells (ciliated cells and Club cells), fibroblasts, endothelial cells and various types of immune cells. The cell cluster map of a representative donor (Asian male, 55-year-old) was visualized using t-distributed stochastic neighbor embedding (tSNE) as shown in Fig. 1b and his major cell type marker expressions were demonstrated in Fig.2.

a. Characteristics of lung transplant donors for single-cell RNA-Seq analysis.
b. Cellular cluster map of the Asian male. All 8 samples were analyzed using the Seurat R package. Cells were clustered using a graph-based shared nearest neighbor clustering approach and visualized using a t-distributed Stochastic Neighbor Embedding (tSNE) plot.

{kind=link}
{kind=link}
AGER, type I alveolar cell marker; SFTPC (SPC), type II alveolar cell marker; SCGB3A2, Club cell marker; TPPP3, ciliated cell marker; CD68, macrophage marker; PTPRC(CD45), pan-immune cell marker.
Next, we analyzed the cell-type-specific expression pattern of ACE2 in each individual. For all donors, ACE2 is expressed in 0.64% of all human lung cells. The majority of the ACE2-expressing cells (averagely 83%) are AT2 cells. Averagely 1.4±0.4% of AT2 cells expressed ACE2. Other ACE2 expressing cells include AT1 cells, airway epithelial cells, fibroblasts, endothelial cells, and macrophages. However, their ACE2-expressing cell ratio is low and variable among individuals. For the representative donor (Asian male, 55-year-old), the expressions of ACE2 and cell-type-specific markers in each cluster are demonstrated in Fig.2.
To further understand the special population of ACE2-expressing AT2, we performed gene ontology enrichment analysis to study which biological processes are involved with this cell population by comparing them with the AT2 cells not expressing ACE2. Surprisingly, we found that multiple viral process-related GO are significantly over-presented, including “positive regulation of viral process” (P value=0.001), “viral life cycle” (P value=0.005), “virion assembly” (P value=0.03) and “positive regulation of viral genome replication” (P value=0.04). These highly expressed viral process-related genes in ACE2-expressing AT2 include: SLC1A5, CXADR, CAV2, NUP98, CTBP2, GSN,HSPA1B,STOM, RAB1B, HACD3, ITGB6, IST1,NUCKS1,TRIM27, APOE, SMARCB1,UBP1,CHMP1A,NUP160,HSPA8,DAG1,STAU1,ICAM1,CHMP5,D EK, VPS37B, EGFR, CCNK, PPIA, IFITM3, PPIB, TMPRSS2, UBC, LAMP1 and CHMP3. Therefore, it seems that the 2019-nCov has cleverly evolved to hijack this population of AT2 cells for its reproduction and transmission.
We further compared the characteristics of the donors and their ACE2 expressing patterns. No association was detected between the ACE2-expressing cell number and the age or smoking status of donors. Of note, the 2 male donors have a higher ACE2-expressing cell ratio than all other 6 female donors (1.66% vs. 0.41% of all cells, P value=0.07, Mann Whitney Test). In addition, the distribution of ACE2 is also more widespread in male donors than females: at least 5 different types of cells in male lung express this receptor, while only 2~4 types of cells in female lung express the receptor. This result is highly consistent with the epidemic investigation showing that most of the confirmed 2019-nCov infected patients were men (30 vs. 11, by Jan 2, 2020).
We also noticed that the only Asian donor (male) has a much higher ACE2-expressing cell ratio than white and African American donors (2.50% vs. 0.47% of all cells). This might explain the observation that the new Coronavirus pandemic and previous SARS-Cov pandemic are concentrated in the Asian area.
Altogether, in the current study, we report the RNA expression profile of ACE2 in the human lung at single-cell resolution. Our analysis suggested that the expression of ACE2 is concentrated in a special population of AT2 which expresses many other genes favoring the viral process. This conclusion is different from the previous report which observed abundant ACE2 not only in AT2, but also in endothelial cells8. In fact, to our knowledge, endothelial cells sometimes can be non-specifically stained in immunohistochemical analysis. The abundant expression of ACE2 in a population of AT2 explained the severe alveolar damage after infection. The demonstration of the distinct number and distribution of ACE2-expressing cell population in different cohorts can potentially identify the susceptible population. The shortcoming of the study is the small donor sample number, and that the current technique can only analyze the RNA level but not the protein level of single cells. Furthermore, it remains unknown whether there is any other co-receptor responsible for the 2019-nCov infection, which might also help to explain the observed difference of transmission ability between SARS-Cov and 2019-nCov. Future work on the ACE2 receptor profiling could lead to novel anti-infective strategies such as ACE2 protein blockade or ACE2-expressing cell ablation.
Methods
Public datasets (GEO: GSE122960) were used for bioinformatics analysis. Firstly, we used Seurat (version 2.3.4) to read a combined gene-barcode matrix of all samples. We removed the low-quality cells with less than 200 or more than 6,000 detected genes, or if their mitochondrial gene content was > 10%. Genes were filtered out that were detected in less than 3 cells. For normalization, the combined gene-barcode matrix was scaled by total UMI counts, multiplied by 10,000 and transformed to log space. The highly variable genes were identified using the function FindVariableGenes. Variants arising from number of UMIs and percentage of mitochondrial genes were regressed out by specifying the vars.to.regress argument in Seurat function ScaleData. The expression level of highly variable genes in the cells was scaled and centered along each gene, and was conducted to principal component analysis.
Then we assessed the number of PCs to be included in downstream analysis by (1) plotting the cumulative standard deviations accounted for each PC using the function PCElbowPlot in Seurat to identify the ‘knee’ point at a PC number after which successive PCs explain diminishing degrees of variance, and (2) by exploring primary sources of heterogeneity in the datasets using the PCHeatmap function in Seurat. Based on these two methods, we selected the first top significant PCs for two-dimensional t-distributed stochastic neighbor embedding (tSNE), implemented by the Seurat software with the default parameters. We used FindClusters in Seurat to identify cell clusters for each sample. Following clustering and visualization with t-Distributed Stochastic Neighbor Embedding (tSNE), initial clusters were subjected to inspection and merging based on the similarity of marker genes and a function for measuring phylogenetic identity using BuildClusterTree in Seurat. Identification of cell clusters was performed on the final aligned object guided by marker genes. To identify the marker genes, differential expression analysis was performed by the function FindAllMarkers in Seurat with Wilcoxon rank sum test. Differentially expressed genes that were expressed at least in 25% cells within the cluster and with a fold change more than 0.25 (log scale) were considered to be marker genes. tSNE plots and violin plots were generated using Seurat.
Acknowledgements
This work was funded by National Key Research Program to W. Zuo (2017YFA0104600), National Science Foundation of China (81770073 to W. Zuo, 81570091 to W. Zuo), Youth 1000 Talent Plan of China to W. Zuo, Tongji University (Basic Scientific Research-Interdisciplinary Fund and 985 Grant to W. Zuo), Shanghai Science and Technology Talents Program (19QB1403100 to W. Zuo), and Shanghai East Hospital Annual Grant to W. Zuo.
No comments:
Post a Comment
Comments always welcome!